
How many stars are there in the night sky? A hundred? A thousand? Ten thousand? A hundred thousand? We could just go to Wikipedia, but that’d be lame. We could also go out tonight (provided the weather is nice) and try to count. It would be an exercise in patience, and ultimately, in futility. Mad respect to those past astronomers who catalogued the sky with little more than their eyes and a piece of paper. Maybe we could select a small part of sky, count the stars, then extrapolate from there. But no, we won’t do any of that today.
Today, we will look at the results of the Hipparcos survey, one of the first, large scale surveys of our galactic neighborhood. More than 118’000 stars have been observed over extended periods of time, and their brightness and parallax were measured. Follow-up studies have blown this initial survey out of the water, with up to billions (!) of objects in the night sky sampled — but that is too much data to go over here. For now, we will simply obtain, parse, and analyze the data from the more humble Hipparcos project, and try to answer the initial question: how many stars are there in the night sky?
The Hipparcos Survey
Hipparcos, or the High Precision Parallax Collecting Satellite, and also a play on the name of the ancient Greek astronomer Hipparcus, was a mission by the European Space Agency with the goal of mapping the motion, brightness, and parallax of more than a hundred thousand stars. The satellite launched in 1989, and for 4 years analyzed the 118’000-something predefined objects. The majority of the stars to be observed were selected based on their apparent magnitude — practically all stars potentially visible to the naked eye were included in that input list.
It took another 4 years to comb through the observational data, and compile a catalogue of the positions, velocities, as well as distances of the studied stars, in addition to a plethora of photometric data. The catalogue can be accessed here (in a plain text format), with an explanatory document here. As you can easily see, it is a mess.
Wrestling the Data into a DataFrame
Let us first inspect the data file:

The columns are separated by the | character, and most relevant data appears to be in its own column. The exception here is the position of the star, which is given by two values (right ascension, or RAdeg, and declination, or DEdeg, both measured in degrees) in a single column. Not shown in the image above, but also present, are the standard deviations of all astrometric data. Likewise, photometric quantities are included for most stars, however, those won’t be relevant for us now.
We thus import the relevant libraries (pandas, numpy, and pyplot, notably):
import pandas as pd import numpy as np from matplotlib import pyplot as plt
And start with reading the input data as a CSV file. We specify the delimiter to be |, skip the first 11 as well as the last row, and tell pandas to ignore the header. Next, we want the parser to only care about specific columns. The list of columns passed to the usecols argument and the list of labels passed to the column names argument should make clear what data we are interested in:
import pandas as pd
import numpy as np
from matplotlib import pyplot as plt
import seaborn as sns
def parse_input_file(filename):
data = pd.read_csv(filename, delimiter='|', engine='python',
skiprows=11,
skipfooter=1,
index_col=0,
header=0,
usecols=[1, 4, 7, 9, 10, 11, 12, 13, 14, 15, 16],
names=['index', 'magnitude', 'raw position', 'parallax',
'proper motion alpha', 'proper motion delta',
'error alpha', 'error delta', 'error parallax',
'error motion alpha', 'error motion delta'])
# convert position string into two floats for the two position coordinates
data = parse_raw_position(data)
# remove rows with NaN values (data missing)
data.dropna(inplace=True)
# convert all miliarcsecond values to degrees
data = convert_to_degrees(data)
# reinterpret magnitude values as floats
data['magnitude'] = data['magnitude'].astype(float)
return data
Once the data is read in, we need to take care of a few issues. First, the position of each star is determined by two numbers present in the same column. We need to take the 'raw position' column and apply the string split function to each entry. We specify expand=True in order to split the original column into several new ones. The values are to be interpreted as floating point numbers instead of strings. Finally, the original column is dropped:
def parse_raw_position(data):
data[['alpha', 'delta']] = (
data['raw position']
.str.split(expand=True)
.astype(float)
)
# drop original column
data.drop('raw position', axis=1, inplace=True)
return data
Next, we convert all input provided in miliarcseconds (mas) to degrees. The relevant columns are multiplied by 1000 to go from miliarcseconds to arcseconds, and then by another factor of 3600 to obtain the values in degrees:
import pandas as pd
import numpy as np
from matplotlib import pyplot as plt
import seaborn as sns
def convert_to_degrees(data):
columns = ['parallax', 'proper motion alpha', 'proper motion delta',
'error alpha', 'error delta', 'error parallax',
'error motion alpha', 'error motion delta']
# divide all miliarcsecond values by 3600 and 1000 to converto to degrees
for column in columns:
data[column] = data[column].astype(float) / 3600000.0
return data
Now that all data entries are of the correct type and have consistent units, we can visually check out the catalogue by looking at some plots:
data = parse_input_file('data.txt')
plt.hist2d(data['alpha'], data['delta'], bins=125, cmap='Blues')
plt.xlabel('ascension (°)')
plt.ylabel('declination (°)')
plt.xticks([0, 60, 120, 180, 240, 300, 360])
plt.yticks([-90, -60, -30, 0, 30, 60, 90])
plt.show()

Interestingly, the stars are not distributed uniformly across the sky. First of all, it is important to realize that the position of stars in the sky is given by two coordinates: right ascension and declination. These are spherical angular coordinates, and thus can not be straightforwardly mapped to a rectangular plot. Hence, the star density in the very north and south (top and bottom, respectively) seems to be lower — but this is just an artifact of the coordinate system used. What is more interesting are the two ‘snakes’ of higher density weaving their way through their plot. The ‘U’ shaped one corresponds, presumably, to the Milky Way, the sideways projection of our Galaxy that appears as a bright band of stars stretching across the night sky. As the Earth’s rotation axis (defining the North and the South poles) is not perpendicular to the Galactic disc, the straight band is warped into a wavy pattern in a ‘geocentric’ coordinate system. However, it is not clear to me what the less apparent ‘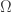
Distance
I was mentioning the position of the stars (in the night sky), as well as their distance. However, till now we only saw parallaxes. What is this parallax? And how do we find the distance of the star from it?
Parallax is the physical phenomenon which, in normal words, is related to the fact that objects appear to be in different places when observed from different angles. Hold an upright finger in front of your face, and close one eye. Then switch eyes without moving the finger — it moves with respect to the distant background. This effect can be used to measure astronomical distances. As the Earth orbits the Sun, it moves in a circle of radius 1 AU (around 1.5 million kilometers). Hence the same observation done half a year later is carried out from a vantage point some 2 AU away. The following graphics illustrate this:




The distance 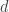


In Python, we can calculate it as follows:
def calculate_distance(data):
data['distance'] = 1.58125e-5 / np.tan(np.pi / 180.0 * data.loc[data['parallax'] > 0.0000001]['parallax'])
return data
data = calculate_distance(data)
Note that we convert the degrees into radians, then multiply by a conversion factor leading to a distance measured in lightyears. We just need to be careful about negative parallaxes, which are, as the program supervisors note, a consequence of measurement errors. Let’s look at the distribution of the stars sampled in the Hipparcos survey as a function of distance:
plt.hist(data['distance'], bins=100)
plt.xlabel('distance (ly)')
plt.ylabel('count')
plt.ylim([0, 10000])
plt.show()

The closest star is indeed Alpha Centauri, located, well, where it is supposed to be located, at a distance of 4.22 lightyears:
data.iloc[data['distance'].argmin()] magnitude 1.101000e+01 parallax 2.145361e-04 proper motion alpha -1.048789e-03 proper motion delta 2.133778e-04 error alpha 3.638889e-07 error delta 4.194444e-07 error parallax 6.722222e-07 error motion alpha 4.222222e-07 error motion delta 5.055556e-07 alpha 2.174489e+02 delta -6.268135e+01 distance 4.223016e+00 Name: 70890, dtype: float64
Visible Stars
The brightness of stars is measured in magnitudes. A magnitude is a number that describes how much brighter or dimmer a given object is with respect to an agreed upon reference. It is a logarithmic scale, and is set up such that a difference of 5 magnitudes corresponds to a factor of 100 in the brightness of the objects. Moreover, somewhat confusingly, brighter objects have a smaller magnitude. And so, in order to filter only the visible stars, all we need to do is to isolate those with a magnitude of around 6.5 and less. A magnitude of 6.5 is on the edge of being seen by a human eye under perfect conditions, and is thus chosen as the cutoff:
def filter_visible(data, cutoff_magnitude):
visible = data[data['magnitude'] < cutoff_magnitude]
return visible
visible = filter_visible(data, cutoff_magnitude=6.5)
We can now plot a subset of the night sky. How about the rectangle of ascension between 10h and 14h, and declination between 40 and 70 degrees? Recognize the pattern of stars?
brightness = np.power(10, -(visible['magnitude'] - visible['magnitude'].min()) / 10)
brightness = (1 - brightness).to_numpy()
plt.scatter(visible['alpha'], visible['delta'], c=brightness, s=brightness * 4,
cmap='Greys', marker='.', vmin=0.5, vmax=1.0)
plt.xlim([220, 140])
plt.ylim([40, 70])
plt.xlabel('ascension (°)')
plt.ylabel('declination (°)')
ax = plt.gca()
ax.set_facecolor((0.0, 0.0, 0.0))
plt.show()

So, How Many Stars?
Let’s look at the number of rows of the visible DataFrame:
visible.info() Int64Index: 8785 entries, 25 to 118322 Data columns (total 11 columns): # Column Non-Null Count Dtype --- ------ -------------- ----- 0 magnitude 8785 non-null float64 1 parallax 8785 non-null float64 2 proper motion alpha 8785 non-null float64 3 proper motion delta 8785 non-null float64 4 error alpha 8785 non-null float64 5 error delta 8785 non-null float64 6 error parallax 8785 non-null float64 7 error motion alpha 8785 non-null float64 8 error motion delta 8785 non-null float64 9 alpha 8785 non-null float64 10 delta 8785 non-null float64 dtypes: float64(11) memory usage: 823.6 KB
8785 entries. But keep in mind, the data span the whole night sky, half of which is obstructed by our planet at any given point in time. This means that there are around 4400 stars visible on a good night. Of course, when the conditions for observation are worse, the number is smaller (often by orders of magnitude in cities). On the other hand side, I only considered stars the average magnitude of which is below the visibility limit. At any given time, there will be many variable stars which may be visible at maximum brightness despite being too dim to observe consistently throughout the year.
Conclusion
That’s it! 4400 stars visible in the night sky. The snake and bears can go to sleep now.
There’s much more data present in the Hipparcos catalogue. For example, the movement of each star was measured, giving us its speed and direction as it travels through the night sky. Obviously, this velocity is infinitesimal, measured in thousandths of arcseconds per year. But over countless millennia, the motion becomes apparent. Next time, we will try to simulate how the night sky will look like in the distant future!
Until next time!

 time complexity and are (nearly) in-place, requiring little to no extra memory space. As I hopefully managed to show, they may look abstract at first, but are very elegant recursive algorithms. However, do they represent the best we can do? Today we will look at some linear time sorting algorithms that perform even faster than heap- or quick sort.
time complexity and are (nearly) in-place, requiring little to no extra memory space. As I hopefully managed to show, they may look abstract at first, but are very elegant recursive algorithms. However, do they represent the best we can do? Today we will look at some linear time sorting algorithms that perform even faster than heap- or quick sort.





 is a vector in a three-dimensional, cartesian space, the integration region is the sphere
is a vector in a three-dimensional, cartesian space, the integration region is the sphere  ,
, 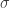 is a real constant, and
is a real constant, and  represents the dielectric constant. In homogeneous and isotropic materials,
represents the dielectric constant. In homogeneous and isotropic materials, 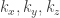 , and if they lie within the sphere
, and if they lie within the sphere 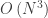 , and already for values around
, and already for values around  scaling, resulting in a runtime of some 4400 seconds! Pretty fast, isn’t it? But we can do even better!
scaling, resulting in a runtime of some 4400 seconds! Pretty fast, isn’t it? But we can do even better!

 . Making the displacement
. Making the displacement 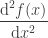
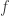 at the three points
at the three points  ,
,  , and
, and  :
:
 away from
away from 

 , which we can index using an integer
, which we can index using an integer 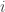 . For each of these values, we can write down an equation of the form:
. For each of these values, we can write down an equation of the form:
 is the total number of points in the discretized domain. Using the approximation of the second derivative shown above, we have that:
is the total number of points in the discretized domain. Using the approximation of the second derivative shown above, we have that: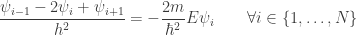

 representing the solution, and a corresponding set of eigenvalues
representing the solution, and a corresponding set of eigenvalues  which represent the appropriately scaled energy levels.
which represent the appropriately scaled energy levels. . This would imply two separate but trivial equations for the values
. This would imply two separate but trivial equations for the values  and
and 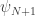 . Moreover, as these values are zero by design, the differential operator is not affected by their presence. Hence, zero boundary conditions can be implemented by simply disregarding the equations for
. Moreover, as these values are zero by design, the differential operator is not affected by their presence. Hence, zero boundary conditions can be implemented by simply disregarding the equations for 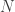 equations inside the discretized domain.
equations inside the discretized domain.
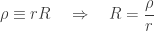
![\dfrac{\partial }{\partial r}\left(r^2\dfrac{\partial }{\partial r}\dfrac{\rho}{r}\right) = \dfrac{\partial }{\partial r}\left[r^2\dfrac{1}{r}\dfrac{\partial \rho}{\partial r} + r^2\rho\left(-\dfrac{1}{r^2}\right)\right] = \dfrac{\partial \rho}{\partial r} + r\dfrac{\partial^2 \rho}{\partial r^2} - \dfrac{\partial \rho}{\partial r} = r\dfrac{\partial^2 \rho}{\partial r^2}](https://s0.wp.com/latex.php?latex=%5Cdfrac%7B%5Cpartial+%7D%7B%5Cpartial+r%7D%5Cleft%28r%5E2%5Cdfrac%7B%5Cpartial+%7D%7B%5Cpartial+r%7D%5Cdfrac%7B%5Crho%7D%7Br%7D%5Cright%29+%3D+%5Cdfrac%7B%5Cpartial+%7D%7B%5Cpartial+r%7D%5Cleft%5Br%5E2%5Cdfrac%7B1%7D%7Br%7D%5Cdfrac%7B%5Cpartial+%5Crho%7D%7B%5Cpartial+r%7D+%2B+r%5E2%5Crho%5Cleft%28-%5Cdfrac%7B1%7D%7Br%5E2%7D%5Cright%29%5Cright%5D+%3D+%5Cdfrac%7B%5Cpartial+%5Crho%7D%7B%5Cpartial+r%7D+%2B+r%5Cdfrac%7B%5Cpartial%5E2+%5Crho%7D%7B%5Cpartial+r%5E2%7D+-+%5Cdfrac%7B%5Cpartial+%5Crho%7D%7B%5Cpartial+r%7D+%3D+r%5Cdfrac%7B%5Cpartial%5E2+%5Crho%7D%7B%5Cpartial+r%5E2%7D&bg=ffffff&fg=333333&s=0&c=20201002)

 we get:
we get:

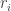 of
of 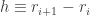 . The discretized Hamiltonian consists of three terms. The first corresponds to the Laplace operator. Using a three-point stencil, we obtain the corresponding tridiagonal matrix:
. The discretized Hamiltonian consists of three terms. The first corresponds to the Laplace operator. Using a three-point stencil, we obtain the corresponding tridiagonal matrix:


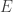 , and the corresponding eigenvectors
, and the corresponding eigenvectors 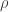 are related to the radial part of the hydrogen wave function
are related to the radial part of the hydrogen wave function 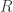 through the simple substitution we adopted earlier,
through the simple substitution we adopted earlier, 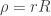 .
. , as well as the matrix corresponding to the discretized Laplace operator. All three functions take the discretized radial coordinate
, as well as the matrix corresponding to the discretized Laplace operator. All three functions take the discretized radial coordinate 
 , we are sampling the hydrogen
, we are sampling the hydrogen  states (
states ( ). The corresponding energies should be
). The corresponding energies should be  , respectively. And that’s exactly what we observe!
, respectively. And that’s exactly what we observe!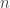 , the permitted values of
, the permitted values of  . Hence, if for instance we choose
. Hence, if for instance we choose  , the lowest energy solution is the one with
, the lowest energy solution is the one with  .
.
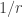 potential. Ideally, either a coordinate transformation, or a an adaptive grid would need to be used to better sample the region close to the nucleus, and not waste grid points far away from it. As it stands now, the
potential. Ideally, either a coordinate transformation, or a an adaptive grid would need to be used to better sample the region close to the nucleus, and not waste grid points far away from it. As it stands now, the 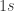 state which is closest to the singularity at the origin is plagued by the largest error. Third, for higher states (like the
state which is closest to the singularity at the origin is plagued by the largest error. Third, for higher states (like the 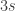 here), increasing the number of points above a certain value does not improve accuracy. This is due to the finite size of the sampled domain. The higher the principal quantum number, the further the electron is from the nucleus. Since we only sample a region of 20 nm, it is too small to completely accommodate higher states.
here), increasing the number of points above a certain value does not improve accuracy. This is due to the finite size of the sampled domain. The higher the principal quantum number, the further the electron is from the nucleus. Since we only sample a region of 20 nm, it is too small to completely accommodate higher states. 
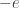 and mass
and mass 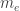 , and a proton of charge
, and a proton of charge  and mass
and mass  . Before we delve into quantum mechanics, we can make one very fundamental approximation. The proton is more than a thousand times heavier than the electron. Hence, it will behave much more like a classical particle than the electron will. So, let us assume the proton is just a classical, charged particle.
. Before we delve into quantum mechanics, we can make one very fundamental approximation. The proton is more than a thousand times heavier than the electron. Hence, it will behave much more like a classical particle than the electron will. So, let us assume the proton is just a classical, charged particle. 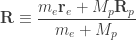
 is the position of the electron and
is the position of the electron and 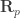 the position of the proton. The center of mass of an isolated system must move at a constant velocity. Therefore, when we express the position of the two particles through the position of the center of mass
the position of the proton. The center of mass of an isolated system must move at a constant velocity. Therefore, when we express the position of the two particles through the position of the center of mass 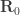 and their relative position
and their relative position  :
:
 must be constant. Knowing this, we can rewrite the two body problem involving two positions
must be constant. Knowing this, we can rewrite the two body problem involving two positions  which is associated with a so-called reduced mass:
which is associated with a so-called reduced mass: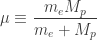
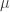 moving in the vicinity of a static particle of opposite charge is determined by the time-dependent Schrödinger equation of motion:
moving in the vicinity of a static particle of opposite charge is determined by the time-dependent Schrödinger equation of motion: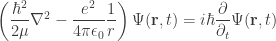


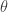 and
and  . The potential written is these coordinates is trivial. The differential operator is a bit more complicated. We can look up the form of the
. The potential written is these coordinates is trivial. The differential operator is a bit more complicated. We can look up the form of the ![\left\{\dfrac{\hbar^2}{2\mu r^2}\left[ \dfrac{\partial}{\partial r}\left(r^2\dfrac{\partial}{\partial r}\right) + \dfrac{1}{\sin\theta}\dfrac{\partial}{\partial\theta}\left(\sin\theta\dfrac{\partial}{\partial\theta}\right) + \dfrac{1}{\sin^2\theta}\dfrac{\partial^2}{\partial\phi^2}\right] - \dfrac{e^2}{4\pi\epsilon_0}\dfrac{1}{r}\right\}\psi =E\psi](https://s0.wp.com/latex.php?latex=%5Cleft%5C%7B%5Cdfrac%7B%5Chbar%5E2%7D%7B2%5Cmu+r%5E2%7D%5Cleft%5B+%5Cdfrac%7B%5Cpartial%7D%7B%5Cpartial+r%7D%5Cleft%28r%5E2%5Cdfrac%7B%5Cpartial%7D%7B%5Cpartial+r%7D%5Cright%29+%2B+%5Cdfrac%7B1%7D%7B%5Csin%5Ctheta%7D%5Cdfrac%7B%5Cpartial%7D%7B%5Cpartial%5Ctheta%7D%5Cleft%28%5Csin%5Ctheta%5Cdfrac%7B%5Cpartial%7D%7B%5Cpartial%5Ctheta%7D%5Cright%29+%2B+%5Cdfrac%7B1%7D%7B%5Csin%5E2%5Ctheta%7D%5Cdfrac%7B%5Cpartial%5E2%7D%7B%5Cpartial%5Cphi%5E2%7D%5Cright%5D+-+%5Cdfrac%7Be%5E2%7D%7B4%5Cpi%5Cepsilon_0%7D%5Cdfrac%7B1%7D%7Br%7D%5Cright%5C%7D%5Cpsi+%3DE%5Cpsi+&bg=ffffff&fg=333333&s=0&c=20201002)
 :
: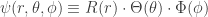
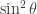 , and divide by the factor in front of the square bracket:
, and divide by the factor in front of the square bracket:![\left[\sin^2\theta\dfrac{\partial}{\partial r}\left(r^2\dfrac{\partial}{\partial r}\right)\! +\! \sin\theta\dfrac{\partial}{\partial\theta}\left(\sin\theta\dfrac{\partial}{\partial\theta}\right)\! + \!\dfrac{\partial^2}{\partial\phi^2}\! - \!\sin^2\theta\dfrac{2\mu r^2}{\hbar^2}\!\left(\dfrac{e^2}{4\pi\epsilon_0}\!\dfrac{1}{r}\! -\! E\right)\!\right]\!\psi\! =\! 0](https://s0.wp.com/latex.php?latex=%5Cleft%5B%5Csin%5E2%5Ctheta%5Cdfrac%7B%5Cpartial%7D%7B%5Cpartial+r%7D%5Cleft%28r%5E2%5Cdfrac%7B%5Cpartial%7D%7B%5Cpartial+r%7D%5Cright%29%5C%21+%2B%5C%21+%5Csin%5Ctheta%5Cdfrac%7B%5Cpartial%7D%7B%5Cpartial%5Ctheta%7D%5Cleft%28%5Csin%5Ctheta%5Cdfrac%7B%5Cpartial%7D%7B%5Cpartial%5Ctheta%7D%5Cright%29%5C%21+%2B+%5C%21%5Cdfrac%7B%5Cpartial%5E2%7D%7B%5Cpartial%5Cphi%5E2%7D%5C%21+-+%5C%21%5Csin%5E2%5Ctheta%5Cdfrac%7B2%5Cmu+r%5E2%7D%7B%5Chbar%5E2%7D%5C%21%5Cleft%28%5Cdfrac%7Be%5E2%7D%7B4%5Cpi%5Cepsilon_0%7D%5C%21%5Cdfrac%7B1%7D%7Br%7D%5C%21+-%5C%21+E%5Cright%29%5C%21%5Cright%5D%5C%21%5Cpsi%5C%21+%3D%5C%21+0&bg=ffffff&fg=333333&s=0&c=20201002)
 :
:

 :
: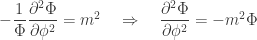
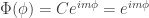
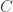 is set to one. The constant
is set to one. The constant 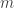 is called the magnetic quantum number. Since any solution must be symmetric with respect to a rotation of 360°, the magnetic quantum number is an integer.
is called the magnetic quantum number. Since any solution must be symmetric with respect to a rotation of 360°, the magnetic quantum number is an integer.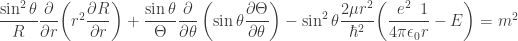
 , and separate the terms that depend on the radial and polar coordinates:
, and separate the terms that depend on the radial and polar coordinates:
 will prove to be a prescient choice. For the polar angle, we find the following equation:
will prove to be a prescient choice. For the polar angle, we find the following equation:

![\dfrac{1}{\sin\theta}\dfrac{\partial }{\partial\theta}\left(\sin\theta\dfrac{\partial \Theta}{\partial\theta}\right) + \left[l(l+1) - \dfrac{m^2}{\sin^2\theta}\right]\Theta= 0](https://s0.wp.com/latex.php?latex=%5Cdfrac%7B1%7D%7B%5Csin%5Ctheta%7D%5Cdfrac%7B%5Cpartial+%7D%7B%5Cpartial%5Ctheta%7D%5Cleft%28%5Csin%5Ctheta%5Cdfrac%7B%5Cpartial+%5CTheta%7D%7B%5Cpartial%5Ctheta%7D%5Cright%29+%2B+%5Cleft%5Bl%28l%2B1%29+-+%5Cdfrac%7Bm%5E2%7D%7B%5Csin%5E2%5Ctheta%7D%5Cright%5D%5CTheta%3D+0&bg=ffffff&fg=333333&s=0&c=20201002)
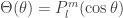 . An interested reader can find a more detailed derivation, for example,
. An interested reader can find a more detailed derivation, for example, 
 :
:
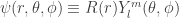
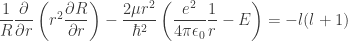
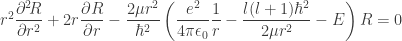
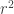 :
:
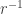 or
or 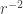 can be neglected, and hence:
can be neglected, and hence:


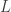 , (b) the principal quantum number
, (b) the principal quantum number 

 is the reduced Bohr radius:
is the reduced Bohr radius: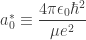
 ,
, is one Rydberg, or 13.605 eV. The energy spectrum is quantized, and follows an inverse square relation. In this solution, which does not take magnetic and spin effects into consideration, solely the principal quantum number
is one Rydberg, or 13.605 eV. The energy spectrum is quantized, and follows an inverse square relation. In this solution, which does not take magnetic and spin effects into consideration, solely the principal quantum number 

 and
and 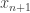 are the current and future population sizes expressed as a fraction of the maximum population, and
are the current and future population sizes expressed as a fraction of the maximum population, and 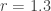 , and try out several starting population sizes (again, expressed as a number between 0 and 1 representing the fraction of the population with respect to the maximum size). The logistic map predicts the following evolution of population size:
, and try out several starting population sizes (again, expressed as a number between 0 and 1 representing the fraction of the population with respect to the maximum size). The logistic map predicts the following evolution of population size:

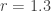 leads to a single asymptotic value. And so do
leads to a single asymptotic value. And so do  and
and  , too. However, starting from a certain value of
, too. However, starting from a certain value of 

 with any starting value
with any starting value 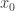 a certain number of times to achieve convergence
a certain number of times to achieve convergence

 evaluated in the algorithm. Likewise, zooming in can be understood as scaling all the values in the complex plane by some factor smaller than one. The end effect will look like we move the camera back and forth, but we won’t need to deal with all the complexities of setting up everything from cameras and projections to model and view transformations.
evaluated in the algorithm. Likewise, zooming in can be understood as scaling all the values in the complex plane by some factor smaller than one. The end effect will look like we move the camera back and forth, but we won’t need to deal with all the complexities of setting up everything from cameras and projections to model and view transformations.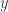 coordinate, respectively, which in our case corresponds to moving along the complex direction. The handling of the left and right arrow keys should be self-explanatory in the light of the above. Note that we scale the “speed” of translation by our
coordinate, respectively, which in our case corresponds to moving along the complex direction. The handling of the left and right arrow keys should be self-explanatory in the light of the above. Note that we scale the “speed” of translation by our 

 , where the initial value of
, where the initial value of  . Take the result of this (let’s call it
. Take the result of this (let’s call it  ) and plug it back into the function. Repeat with
) and plug it back into the function. Repeat with 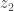 ,
, 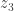 ,
,  ad infinitum. Is the modulus of
ad infinitum. Is the modulus of  , its distance from the origin of the complex plane, below
, its distance from the origin of the complex plane, below  ? If yes, then
? If yes, then  :
:




 has a modulus of
has a modulus of 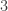 , and
, and  , hence
, hence  is not in the Mandelbrot set.
is not in the Mandelbrot set. ? Then we get the series
? Then we get the series  ,
,  ,
,  ,
,  . And since this number is smaller than
. And since this number is smaller than 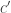 , infinitesimally larger or smaller, might take ten billion iterations of the process before
, infinitesimally larger or smaller, might take ten billion iterations of the process before  .
. 
 then stop and return
then stop and return  go to 1. and repeat
go to 1. and repeat in size and centered around zero (with a slight shift in the negative real direction).
in size and centered around zero (with a slight shift in the negative real direction).


 array, to hold the values of the electrostatic potential on the real space grid.
array, to hold the values of the electrostatic potential on the real space grid. being oriented perpendicular to the surface. As
being oriented perpendicular to the surface. As 


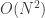 with the size
with the size 








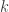 smallest values present in an array of length
smallest values present in an array of length  for the
for the 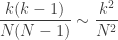 in the case of
in the case of 